

-
製剤写真・
組成・性状 - お知らせ
- コード・番号表
- FAQ
- 電子添文
- インタビューフォーム
-
くすりのしおり
(日本語) -
くすりのしおり
(英語) - 適正使用ガイド
- RMP
-
患者向
医薬品ガイド - 製造販売後調査
- 患者指導箋
-
その他の
適正使用資料
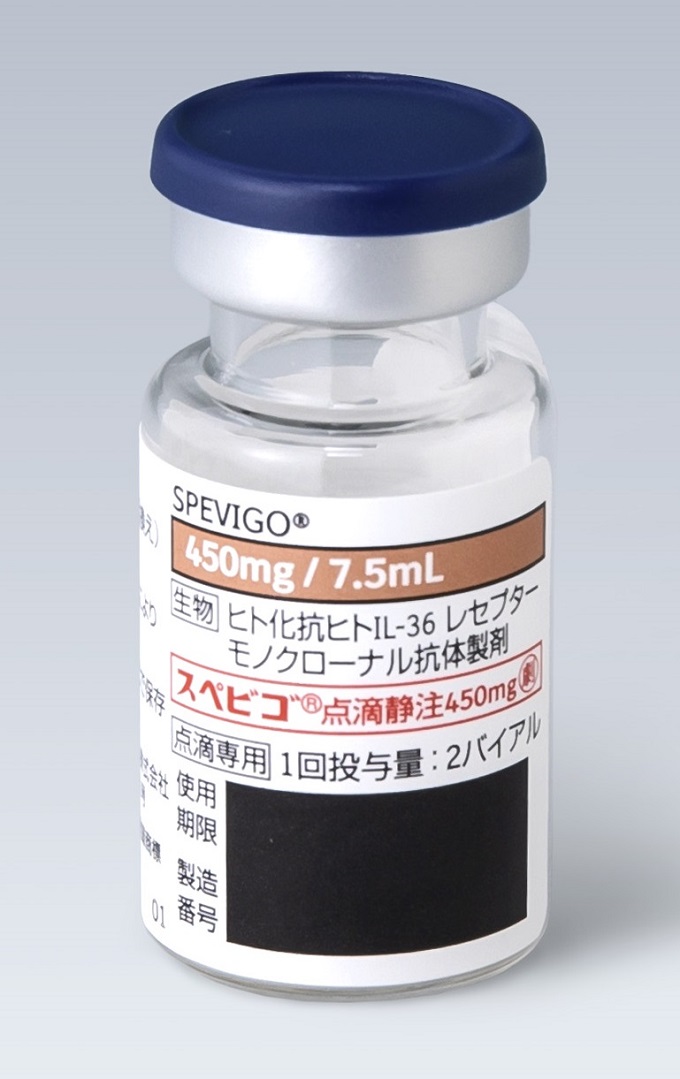
製剤写真・組成・性状
組成・性状
| 成分名 | スペソリマブ(遺伝子組換え) |
|---|---|
| 領域名 | 皮膚・炎症性・免疫性疾患 |
| 成分・含量 | 1バイアル7.5mL中 スペソリマブ(遺伝子組換え)450mg |
| 剤形・色調 | 注射剤(バイアル) |
| 性状 | 無色~微黄褐色の澄明又はわずかに乳白光を呈する液 |
| pH | 5.2~5.8 |
包装形態
コード・番号表
| 日本標準商品分類番号 | 873999 |
|---|---|
| 承認番号 | 30400AMX00409000 |
| 薬価基準収載 医薬品コード |
3999466A1026 |
| レセプト電算処理コード | 629920901 |
包装:7.5mL[2バイアル]
| JANコード | 4987413064118 |
|---|---|
| HOT7(処方用番号) | 1992090 |
| HOT番号 | 1992090010101 |
| 販売包装単位コード | (01)14987413064115 |
| 調剤包装単位コード | (01)04987413951135 |
よくある質問
-
本剤はペプチド及びアミノ酸に代謝されると推定されます。
<引用>
スペビゴ点滴静注 電子添文 -
本剤を生理食塩液以外に希釈したデータは無く、安定性が不明のため生理食塩液に希釈していただくようお願い致します。
<参考>
本剤は希釈前に目視による確認を行い、濁り、異物又は変色が認められる場合には使用しないこと。
日局生理食塩液100mL点滴バッグ又はボトルから15mLを抜き取る。本剤15mL(2バイアル分)を採取して点滴バッグ又はボトルへ緩徐に注入し、使用前に穏やかに混和すること。
調製した希釈液は速やかに使用すること。<引用>
スペビゴ点滴静注 電子添文 -
本剤と他剤との配合変化を検討したデータはありませんので他剤との配合は避けていただくようお願い致します。
<引用>
スペビゴ点滴静注 インタビューフォーム Ⅳ.製剤に関する項目 8.他剤との配合変化(物理化学的変化) -
本剤の電子添文にも記載されている通り、必要です。
<参考>
本剤と他の薬剤を同一の投与ラインにより連続注入する場合には、本剤の投与前後に生理食塩液を輸液チューブ内に流して洗浄すること<引用>
スペビゴ点滴静注 電子添文 -
本剤の電子添文に記載されている通り、必ずフィルター(無菌、パイロジェンフリーでタンパク結合性の低い0.2 μmインラインフィルター)を使用して投与して下さい。
使用していないことに途中で気づいた場合、直ちに投与を中断しフィルターをつけて再投与して下さい。
バイアルの保存中や希釈液調製時に製剤成分に由来する微粒子が形成される可能性がありますが、これらはフィルターにより除去されます。
適正使用ガイド
情報をシェアする